Trotz neuer Therapieentwicklungen, zu denen auch das Institut für Klinische Neurobiologie beigetragen hat, sind die Mechanismen, die für die Degeneration der axonalen Fortsätze und für die Denervation der Muskelfasern verantwortlich sind, noch weitgehend unbekannt.
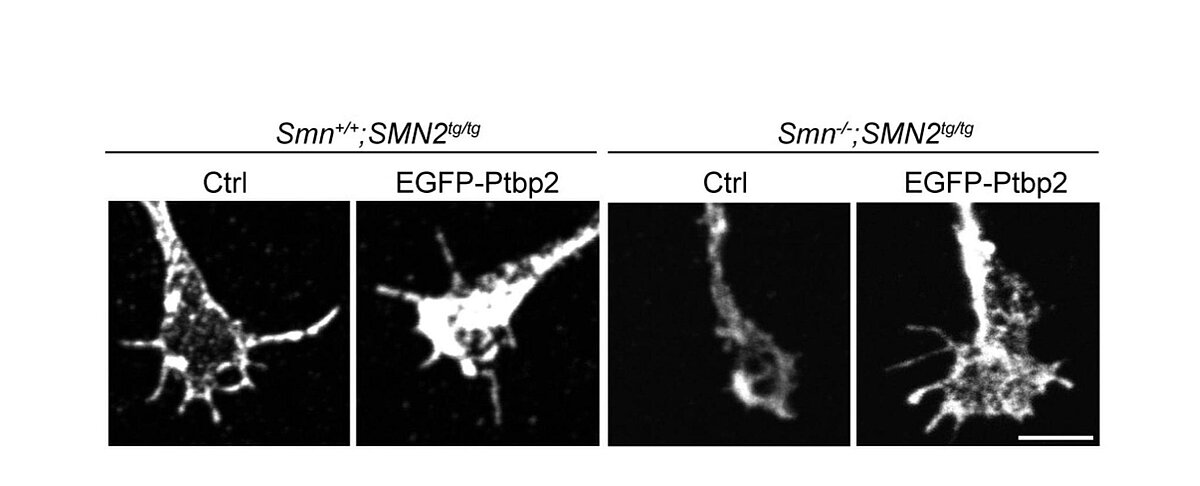
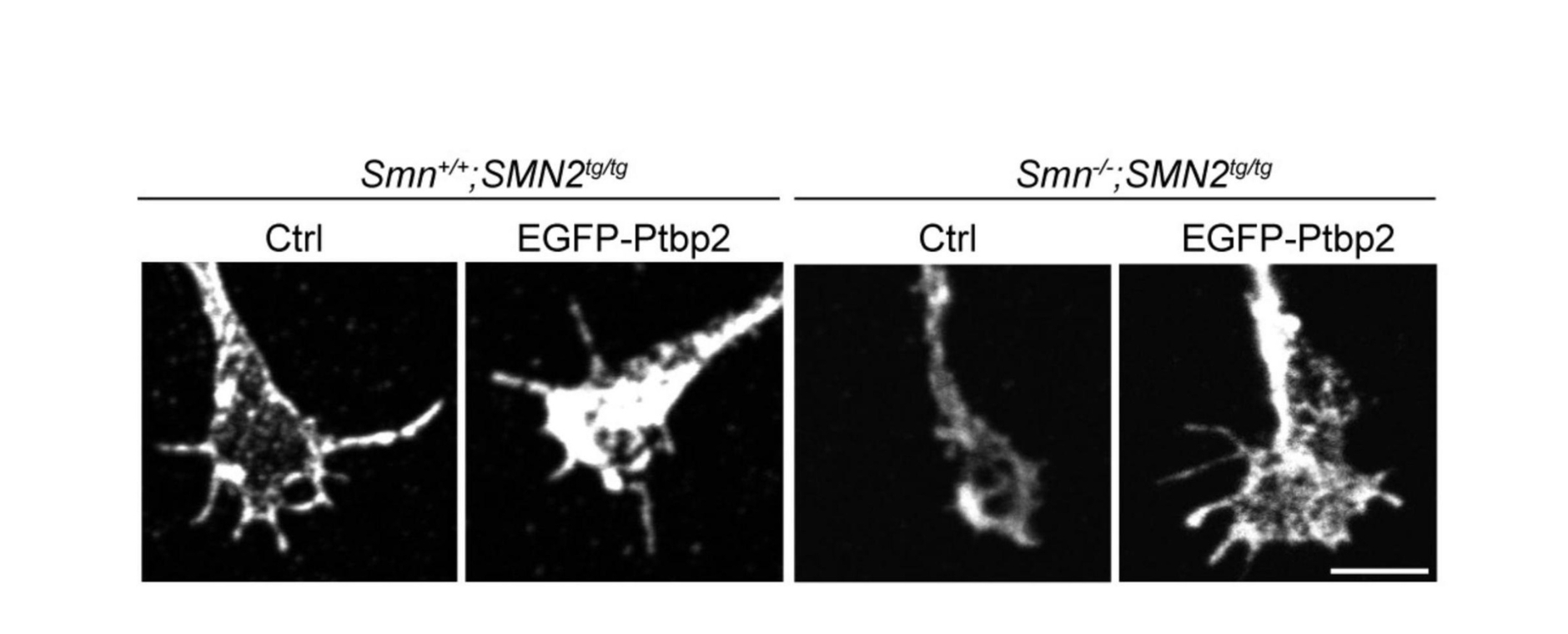
In dieser Arbeit konnte gezeigt werden, dass in einem Mausmodell für spinale Muskelatrophie die Expression des RNA-bindenden Proteins Ptbp2 in den axonalen Fortsätzen von Motoneuronen reduziert ist. Dieses Protein ist wichtig für das Wachstum und die Funktion dieser „Verbindungen“ zwischen Nervenzellen und Muskeln. Wird Ptbp2 wieder zugeführt, können die gestörten Prozesse repariert werden: Die Axone wachsen wieder normal in die Länge, und auch die fehlerhafte Entwicklung der Kontaktstellen, an denen die Nervenzellen Signale weiterleiten, wird verbessert.
Die Ergebnisse zeigen, dass SMN und Ptbp2 gemeinsam die Produktion von Proteinen in den Axonen steuern, die für die Funktion der Nervenzellen entscheidend sind. Daraus könnten sich neue Ansätze für die Behandlung der spinalen Muskelatrophie ergeben.
Publikation:
Salehi Saeede, Zare Abdolhossein, Gandhi Gayatri, Sendtner Michael, Briese Michael. Ptbp2 re-expression rescues axon growth defects in Smn-deficient motoneurons. Front Mol Neurosci. 2024 Aug 23;17:1393779. doi: 10.3389/fnmol.2024.1393779. PMID: 39246602; PMCID: PMC11377325.